

New research from Prof. Vincent Timmerman’s lab, in collaboration with researchers from the University of Oxford, has elucidated the structural changes caused by a mutation in the molecular chaperone Hsp27 (HSPB1). Chaperones are a group of proteins that have functional similarity and assist in protein folding. This work not only sheds light on an important disease-causing mutation, it also reveals an important mechanism by which this chaperone functions.
During their lifetime, cells continuously produce proteins. While many of these proteins contain the correct shape and size, some may contain small errors. To prevent these faulty proteins from causing trouble, the cell makes use of protein quality control factors, such as heat shock proteins. Small heat shock proteins are the smallest of the heat shock protein family, as suggested by their name, and are the first responders to protein misfolding. Their role is to prevent misfolded proteins, such as those responsible for Parkinson’s and Alzheimer’s disease, from accumulating and dysregulating other cellular processes.
Timmerman’s lab has a long-standing interest in understanding Charcot-Marie-Tooth (CMT) disease, the most frequent inherited disorder of the peripheral nervous system. CMT is a genetic nerve disease with over 100 known genetic causes. Onset can be at birth or later in life and is characterised by degeneration of motor nerves and loss of sensation in the feet, hands, legs and arms. It is a progressive disease that can lead to severe disability or even death.
Timmerman’s lab discovered that mutations in the small heat shock proteins Hsp27 (HSPB1) and Hsp22 (HSPB8) lead to CMT disease. While they normally prevent other proteins from misfolding, some mutations cause these chaperones to aggregate themselves. One of the best-known examples is the P182L mutation, located in the intrinsically disordered C-terminal region of HSP27, and which causes a particularly severe form of CMT. The underlying changes in the structure, leading to this misfolding, remained unknown.
In collaboration with Dr. Reid Alderson, Prof. Justin Benesch and Prof. Andrew Baldwin at the University of Oxford, Dr. Elias Adriaenssens (a postdoctoral fellow in the Timmerman lab) worked out how this P182L mutation causes changes to the structure of Hsp27. Starting with in vitro experiments, they characterized the size of the molecular complexes that appear as aggregates inside cells. They found that due to this mutation, these oligomeric complexes can be as large as 14 MDa, the largest form of a small heat shock protein complex ever observed. The researchers then used state-of-the-art microscopy methods, such as expansion microscopy, to confirm that these complexes also appear in human cells and in motor neurons derived from CMT patients using the induced pluripotent stem cell technology.

Using nuclear magnetic resonance (NMR) the changes in structure were characterized at the atomic level and revealed that reduced self-binding of the C-terminal domain to the alpha-crystallin domain leaves a binding pocket more available to other proteins. This was an unexpected finding because the mutated amino acid does not interact directly with the structured core domain. The researchers found that instead it holds its neighbouring amino acids in place to bind to the binding pocket on the structured core domain. Due to this mutation, the binding pocket is less frequently occupied.
To understand the consequences of increased exposure of the binding pocket, the research teams performed protein-protein interaction studies and were able to identify a list of proteins that bind more to this mutant form of HSP27. In an attempt to understand why this specific set of proteins would bind more to the exposed binding pocket, the researchers noticed that many of these proteins contained a similar binding motif to the one in the C-terminal domain of HSP27, but which is now mutated by the P182L mutation. This suggested a new model where proteins with a similar binding motif would compete with the C-terminal domain of HSP27 to bind a hydrophobic binding pocket on the surface of the structured core domain. Dysregulation of this gatekeeper process thus not only leads to changes in the size of HSP27, but it also dysregulates its interactions with other proteins.
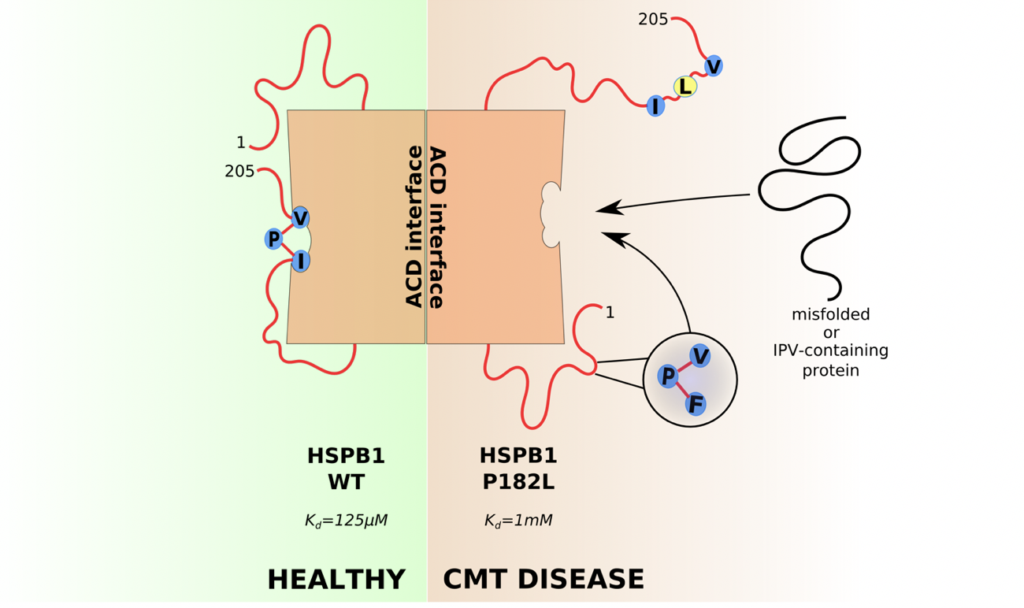
These insights could form an important step towards the development of a therapy for CMT disease caused by mutations in small heat shock proteins.
This work was published in the EMBO Journal and is the start of a productive collaboration between the UAntwerp and Oxford labs. You can find the full paper here: https://www.embopress.org/doi/full/10.15252/embj.2019103811
Elias Adriaenssens Bio
Elias obtained an M.Sc. in Pharmacology (University of Oxford) and an M.Sc. in Biochemistry & Biotechnology (University of Antwerp). He conducted his PhD research in Prof. Vincent Timmerman’s lab, where he studied small heat shock proteins. His research was focused on gaining a better understanding of the structure and function of small heat shock proteins, both in health and disease. The latter with the goal to find a therapy for small heat shock protein associated forms of Charcot-Marie-Tooth disease. Now, Elias is doing his Postdoctoral training with Prof. Sascha Martens at the Max Perutz labs in Vienna (Austria).
The Timmerman lab is a member of the µNEURO Research Centre of Excellence and the iMARK valorisation consortium of the University of Antwerp. The lab is supported by the Research Foundation Flanders (FWO), American Muscular Dystrophy Association (MDA), Medical Foundation Queen Elisabeth (GSKE), Association Belge contre les Maladies Neuromusculaires (ABMM), Rotary ‘Hope in Head’ program and EU-H2020 Solve-RD program ‘Solving the unsolved rare diseases’. Elias Adriaenssens holds an FWO Postdoctoral scholarship.
Article written by Dr. Elias Adriaenssens and edited by Dr. Bronwen Martin.